|
||||||||
| HOME | METHODS | LINKS | NEWS | TIPS | SOFTWARE | FORUM | CONTACT | SEARCH |
| Analysis of DNA methylation using bisulphite sequencing | |
|
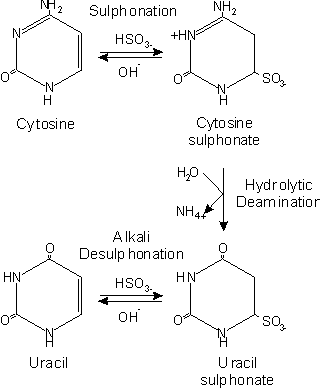
This method allows precise analysis of methylation in a certain region by converting all nonmethylated cytosines into tymines, while methylated cytosines remain unchanged. This method requires small amount of genomic DNA and therefore seems to be very useful for the analysis of clinical samples, where the material amount is limited. However I suggest optimising the method using genomic DNA from a cell line and then apply it to valuable samples. Before starting the experiment you have to develop primers for bisulphite converted DNA. You can generate a model of bisulphite treated DNA by substituting all cytosines which are not in CG context into tymines. And then design your primers in the way that they don not contain any CG. If this is impossible, you have to use C/T at the place of C in CG context. Usually primer selection is the most critical in bisulphite based methylation analysis, since the complexity of DNA is reduced. Therefore I would suggest to select 2-3 pairs of primers, check them on bisulphite modified DNA, and use the most specific ones.
Protocol (Better protocol is available here!)
Obtained PCR product can be sequenced directly, in this case you will obtain more or less reliable results about the percentage of methylated cytosine in every position. Another possibility is to clone the product and then sequence 10 or more individual clones. This method is much more time consuming. Third method which can be used for the analysis of bisulphite treated DNA is Single Nucleotide Primer Extension (SNuPE).
|
|
| \METHODS\DNA methylation analysis\Analysis of methylation using bisulphite sequencing | |
| © Copyright 1999-2006 Alexei Gratchev. All rights reserved. | |